Bioinformatics Strategies for Genome-Wide Association Studies (GWAS)
Our new review on bioinformatics strategies for GWAS analysis has been published in Bioinformatics. We focus in this paper on methods that are designed to embrace, rather than ignore, the complexity of common human diseases.
Moore, J.H., Asselbergs, F.W., Williams, S.M. Bioinformatics strategies for genome-wide association studies. Bioinformatics (2010). [PDF]
ABSTRACT
Motivation: The sequencing of the human genome has made it possible to identify an informative set of more than one million single nucleotide polymorphisms (SNPs) across the genome that can be used to carry out genome-wide association studies (GWAS). The availability of massive amounts of GWAS data has necessitated the development of new biostatistical methods for quality control, imputation, and analysis issues including multiple testing.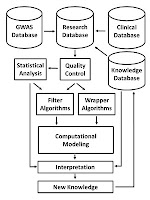 This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving healthcare through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype-phenotype relationship that is characterized by significant heterogeneity and gene-gene and gene-environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving healthcare through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype-phenotype relationship that is characterized by significant heterogeneity and gene-gene and gene-environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
ABSTRACT
Motivation: The sequencing of the human genome has made it possible to identify an informative set of more than one million single nucleotide polymorphisms (SNPs) across the genome that can be used to carry out genome-wide association studies (GWAS). The availability of massive amounts of GWAS data has necessitated the development of new biostatistical methods for quality control, imputation, and analysis issues including multiple testing.
 This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving healthcare through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype-phenotype relationship that is characterized by significant heterogeneity and gene-gene and gene-environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
This work has been successful and has enabled the discovery of new associations that have been replicated in multiple studies. However, it is now recognized that most SNPs discovered via GWAS have small effects on disease susceptibility and thus may not be suitable for improving healthcare through genetic testing. One likely explanation for the mixed results of GWAS is that the current biostatistical analysis paradigm is by design agnostic or unbiased in that it ignores all prior knowledge about disease pathobiology. Further, the linear modeling framework that is employed in GWAS often considers only one SNP at a time thus ignoring their genomic and environmental context. There is now a shift away from the biostatistical approach toward a more holistic approach that recognizes the complexity of the genotype-phenotype relationship that is characterized by significant heterogeneity and gene-gene and gene-environment interaction. We argue here that bioinformatics has an important role to play in addressing the complexity of the underlying genetic basis of common human diseases. The goal of this review is to identify and discuss those GWAS challenges that will require computational methods.
posted by Jason H. Moore, Ph.D. @ 12:36 PM
![]()
![]()


0 Comments:
Post a Comment
<< Home